spatial-smooth: a tutorial¶
⚠️ This package is for looking, not for measuring
spatial-smoothmakes spatial regions easier to see. What it produces is a picture.Smoothing works by making each cell look more like its neighbours. That is exactly what you want when you are trying to spot where a gene programme is switched on — and exactly what you must not feed into a statistical test. Once cells have been made to resemble their neighbours they are no longer independent measurements, so differential expression, cluster comparisons, correlations and p-values computed on smoothed values are badly over-confident. They will report strong, convincing structure in data that contains none.
Every call writes the unsmoothed score next to the smoothed one, as
adata.obs["<name>_raw"]. Look at the smoothed one. Do your statistics on the raw one.
A per-cell signature score is noisy — each cell is measured independently, so dropout and sampling variance dominate, and a real anatomical region can be genuinely hard to pick out of the speckle. Smoothing lets neighbouring cells borrow statistical strength. The scientific choice is which neighbours count:
smoothing |
neighbours are… |
recovers |
|---|---|---|
spatial |
physically adjacent cells |
tissue architecture: niches, layers, gradients |
cell state |
transcriptionally similar cells |
biology, independent of position |
both, composed |
first the manifold, then the tissue |
denoised expression laid out in space |
This notebook walks three levels of control:
One line. Defaults do everything.
Parameterized. Choose the pipeline; forward plotting kwargs to scanpy/squidpy.
Fully modular. Compute → store → write to disk → reload → plot without recomputing.
The data is a public 10x Genomics Xenium mouse-brain section (CC-BY), downloaded on first run (~4.5 MB). Nothing here needs a cluster.
Setup¶
pip install "spatial-smooth[all]"
%matplotlib inline
import numpy as np
import pandas as pd
import scanpy as sc
import spatial_smooth as ss
1. The data¶
Using your own data instead¶
spatial-smooth needs exactly two things from an AnnData, and nothing else:
adata.X(or a layer you name vialayer=) holding log-normalised expression, andadata.obsm["spatial"]holding the cells’ physical coordinates, as an(n_obs, 2)array.
So if you already have a prepared object, skip the download entirely — this is the whole of section 1 for you:
import anndata as ad
adata = ad.read_h5ad("my_section.h5ad")
assert "spatial" in adata.obsm # (n_obs, 2) coordinates
# adata.X must be log-normalised; if it holds raw counts:
# adata.layers["counts"] = adata.X.copy()
# sc.pp.normalize_total(adata); sc.pp.log1p(adata)
Then jump to section 2. Cell-state smoothing (steps="dm") additionally wants
obsm["DM_EigenVectors"], which ss.smooth(..., auto_embed=True) computes for you if absent.
The example dataset¶
The rest of this notebook uses a public 10x Xenium mouse-brain coronal subset: ~36,000 cells, a
248-gene panel, one cell per row with physical centroids. We fetch the two small loose outputs
(cached, so re-running is free), assemble an AnnData, and put the centroids in
obsm["spatial"].
import pathlib, urllib.request
BASE = ("https://cf.10xgenomics.com/samples/xenium/1.0.2/"
"Xenium_V1_FF_Mouse_Brain_Coronal_Subset_CTX_HP")
NAME = "Xenium_V1_FF_Mouse_Brain_Coronal_Subset_CTX_HP"
DATA = pathlib.Path("data/xenium_mousebrain")
DATA.mkdir(parents=True, exist_ok=True)
# Point this at your own .h5ad to run the whole notebook on your data instead.
PREPARED = pathlib.Path("data/prepared.h5ad")
for fname in (f"{NAME}_cell_feature_matrix.h5", f"{NAME}_cells.csv.gz"):
dest = DATA / fname
if PREPARED.exists():
break
if not dest.exists():
print(f"downloading {fname} ...")
urllib.request.urlretrieve(f"{BASE}/{fname}", dest)
print(f" {fname} ({dest.stat().st_size / 1e6:.1f} MB)")
Xenium_V1_FF_Mouse_Brain_Coronal_Subset_CTX_HP_cell_feature_matrix.h5 (2.9 MB)
Xenium_V1_FF_Mouse_Brain_Coronal_Subset_CTX_HP_cells.csv.gz (1.7 MB)
import anndata as ad
if PREPARED.exists():
# --- alternative path: load an object you prepared earlier -------------------
adata = ad.read_h5ad(PREPARED)
print(f"loaded {PREPARED}")
else:
# --- example path: assemble the public Xenium section ------------------------
adata = sc.read_10x_h5(DATA / f"{NAME}_cell_feature_matrix.h5")
adata.var_names_make_unique()
cells = pd.read_csv(DATA / f"{NAME}_cells.csv.gz").set_index("cell_id")
cells.index = cells.index.astype(str)
adata.obs_names = adata.obs_names.astype(str)
adata.obs = adata.obs.join(cells, how="left")
adata.obsm["spatial"] = adata.obs[["x_centroid", "y_centroid"]].to_numpy()
sc.pp.filter_cells(adata, min_counts=10)
adata.layers["counts"] = adata.X.copy()
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
# The only two preconditions, checked explicitly.
assert "spatial" in adata.obsm, "spatial-smooth needs obsm['spatial']"
assert adata.X.max() < 100, "adata.X should be log-normalised, not raw counts"
print(f"{adata.n_obs:,} cells x {adata.n_vars} genes")
36,419 cells x 248 genes
A small hippocampal program from the panel — dentate-gyrus and CA markers. Four genes, each sparse and noisy on its own.
HIPPOCAMPUS = ["Prox1", "Neurod6", "Wfs1", "Fibcd1"]
assert set(HIPPOCAMPUS) <= set(adata.var_names)
HIPPOCAMPUS
['Prox1', 'Neurod6', 'Wfs1', 'Fibcd1']
2. Level one — one line, defaults do everything¶
ss.smooth with no steps argument smooths over obsm["spatial"] with a Gaussian kernel across
each cell’s 400 nearest spatial neighbours. The bandwidth is inferred from the data (six median
nearest-neighbour distances), so you do not pick a number in microns.
ss.pl.signature then plots the raw score next to the smoothed one.
ss.smooth(adata, HIPPOCAMPUS, "hippocampus")
ss.pl.signature(adata, "hippocampus")
WARNING: Please specify a valid `library_id` or set it permanently in `adata.uns['spatial']`
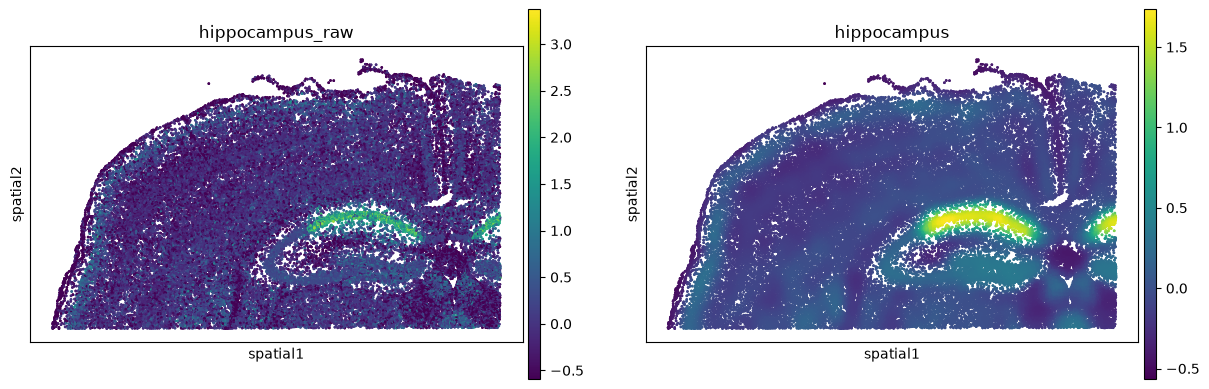
The raw panel is a speckle of individual cells; the smoothed panel resolves the dentate-gyrus
C-shape, the CA fields, and the cortical layers. Two columns appeared in obs, and a record of
what was run in uns.
print(adata.obs[["hippocampus_raw", "hippocampus"]].describe().T)
print()
prov = ss.provenance(adata, "hippocampus")
print("genes :", prov["genes"])
print("score :", prov["score"])
print("pipeline :", [s["kind"] for s in prov["steps"]])
res = prov["steps"][0]["resolved"]
print("bandwidth:", round(res["sigma_used"], 1), "um nominal;",
round(res["sigma_effective"], 1), "um effective",
f"({res['kernel_mass_retained']:.0%} of the kernel kept)")
count mean std min 25% \
hippocampus_raw 36419.0 -3.351830e-09 0.488344 -0.592023 -0.302019
hippocampus 36419.0 5.307676e-03 0.308730 -0.564868 -0.141658
50% 75% max
hippocampus_raw -0.051674 0.205903 3.372188
hippocampus -0.059440 0.044997 1.734469
genes : ['Prox1', 'Neurod6', 'Wfs1', 'Fibcd1']
score : mean_z
pipeline : ['knn_gaussian']
bandwidth: 78.2 um nominal; 71.4 um effective (96% of the kernel kept)
3. Level two — choose the pipeline, control the plot¶
3a. Composition: spatial, cell state, or both¶
steps selects what you smooth over:
|
pipeline |
meaning |
|---|---|---|
|
|
spatial only |
|
|
cell state only |
|
|
both, cell state first |
Doing just one of the two is the ordinary case, not a special one — a one-element pipeline. Composing runs the steps left to right: the spatial step smooths the expression the cell-state step already denoised.
The cell-state step is a Gaussian-process regression over a diffusion map of the expression
manifold (kompot.smooth_expression, built on mellon). It needs obsm["DM_EigenVectors"];
with auto_embed=True (the default) spatial-smooth computes it with Palantir if absent.
Everything below runs on the full section — every cell, no subsampling. The Gaussian process is the slow step (a few minutes); the two spatial smoothers take about a second each.
ss.compute_diffusion_map(adata) # Palantir -> obsm["DM_EigenVectors"]
adata.obsm["DM_EigenVectors"].shape
(36419, 10)
# cell state only: GP over the diffusion map
ss.smooth(adata, HIPPOCAMPUS, "dm_only", steps="dm")
# spatial only: Gaussian kNN over tissue coordinates
ss.smooth(adata, HIPPOCAMPUS, "spatial_only", steps="spatial")
# both, composed: manifold first, then tissue
ss.smooth(adata, HIPPOCAMPUS, "composed", steps="dm+spatial")
ss.list_results(adata)
[2026-07-09 21:55:19,898] [INFO ] Smoothing all 36,419 cells
[2026-07-09 21:58:18,553] [INFO ] Smoothing all 36,419 cells
['composed', 'dm_only', 'hippocampus', 'spatial_only']
ss.pl.compare(
adata, ["spatial_only", "dm_only", "composed"], raw=True,
backend="scanpy", ncols=4, frameon=False,
)
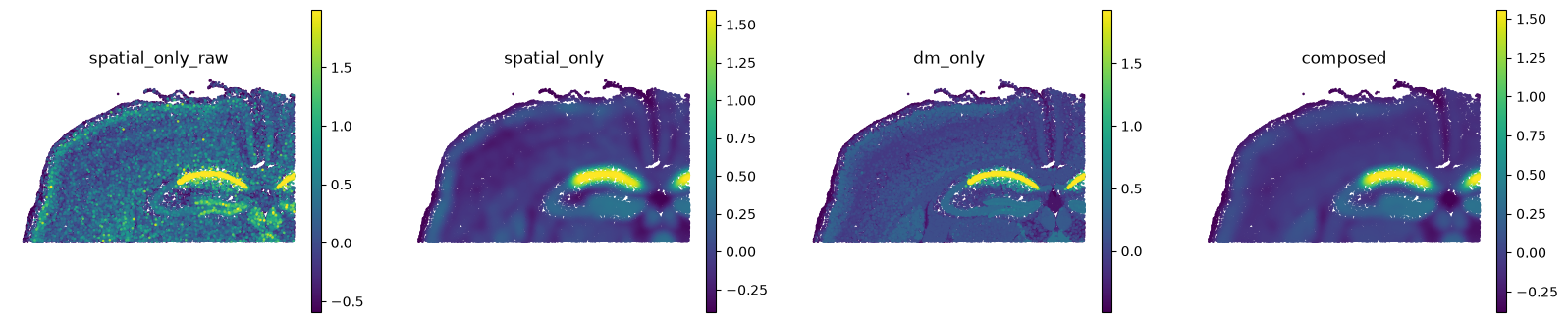
Read the four panels left to right: the raw score, then each pipeline. Spatial smoothing produces the cleanest tissue field. Cell-state smoothing denoises without using position at all. Composing does both, and is the smoothest of the three.
3b. Plot control: kwargs go straight through¶
ss.pl.signature is a wrapper, not a reimplementation. Everything after name is forwarded
verbatim to the backend:
|
underlying call |
|---|---|
|
|
|
|
|
|
|
squidpy if installed, else scanpy |
color is set for you from the stored provenance. Defaults (cmap, percentile colour limits,
a grey na_color) are injected only for keys you did not pass.
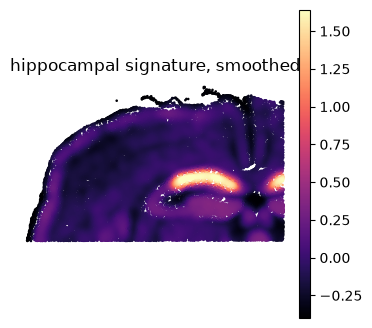
ss.pl.signature(
adata, "hippocampus", raw=False,
backend="scanpy", # -> scanpy.pl.embedding
cmap="magma", vmax="p99.5", frameon=False,
title="hippocampal signature, smoothed",
)
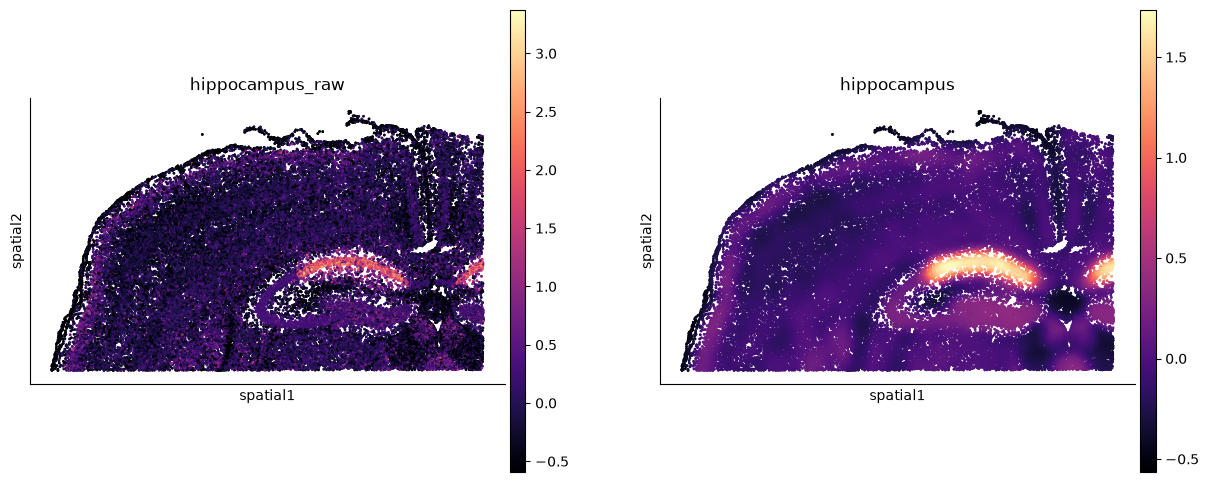
# The same result through squidpy, which knows about tissue images and library ids.
ss.pl.signature(adata, "hippocampus", backend="squidpy", cmap="magma", figsize=(6, 6))
WARNING: Please specify a valid `library_id` or set it permanently in `adata.uns['spatial']`
3c. Bandwidth is scale-invariant¶
Every default bandwidth is a multiple of the median nearest-neighbour distance, so the same factor smooths the same amount whether coordinates are microns or millimetres. Rescale the coordinates a thousandfold and the field is unchanged.
rescaled = adata.copy()
rescaled.obsm["spatial"] = rescaled.obsm["spatial"] * 1000.0
ss.smooth(rescaled, HIPPOCAMPUS, "hippocampus")
a = adata.obs["hippocampus"].to_numpy()
b = rescaled.obs["hippocampus"].to_numpy()
ra = ss.provenance(adata, "hippocampus")["steps"][0]["resolved"]
rb = ss.provenance(rescaled, "hippocampus")["steps"][0]["resolved"]
print("max |difference| :", np.abs(a - b).max())
print("sigma_effective (um) :", round(ra["sigma_effective"], 2))
print("sigma_effective (nm) :", round(rb["sigma_effective"], 2))
print("(nominal was", round(ra["sigma_nominal"], 2), "um -- the number NOT to quote)")
max |difference| : 0.0
sigma_effective (um) : 71.45
sigma_effective (nm) : 71448.38
(nominal was 78.15 um -- the number NOT to quote)
One caveat worth internalising. The Gaussian process infers its length scale the same way, via
ls_factor. Over a diffusion map kompot’s nativels_factor=10is right; over physical coordinates it is ~200x the cell spacing and washes the field into a single global gradient. Usels_factor≈0.3there — which is exactly what the"spatial-gp"shorthand does.
4. Level three — fully modular: compute, store, plot later¶
Pass Step objects instead of a shorthand for complete control. Each step is a frozen dataclass:
a specification, not a fitted object, so it can be reused and is recorded verbatim.
pipeline = [
ss.KompotGP(basis="DM_EigenVectors", ls_factor=10.0, n_landmarks=5000),
ss.KnnGaussian(basis="spatial", k=64, sigma_factor=4.0),
]
pipeline
[KompotGP(basis='DM_EigenVectors', sigma=1.0, ls=None, ls_factor=10.0, n_landmarks=5000, groupby=None, condition=None, random_state=0),
KnnGaussian(basis='spatial', k=64, sigma=None, sigma_factor=4.0, workers=-1)]
# k=64 truncates the Gaussian. The package says so, on stderr, and the notebook keeps that
# message: it is the disclosure this section exists to teach.
ss.smooth(adata, HIPPOCAMPUS, "custom", steps=pipeline, store_genes=True)
print("smoothed score :", adata.obs["custom"].shape)
print("smoothed expression :", adata.obsm["custom_smoothed"].shape) # store_genes=True
[2026-07-09 22:01:06,232] [INFO ] Smoothing all 36,419 cells
smoothed score : (36419,)
smoothed expression : (36419, 4)
steps.py:191: TruncationWarning: KnnGaussian(k=64) truncates the kernel: only 69% of the Gaussian mass falls within each point's 64-neighbour radius, so the effective bandwidth is 35.7 (nominal sigma 52.1) and varies with local density. Raise k, or quote the effective bandwidth, not the nominal one.
W, sigma_used, info = knn_gaussian_operator(
Read the warning — it is doing its job¶
That pipeline chose k=64, and the package objected. Restricting the Gaussian to a cell’s 64
nearest neighbours cuts the kernel off before it has faded: only ~69% of its weight lies
inside that radius. The bandwidth the data actually feels is therefore narrower than the
nominal sigma, and — because 64 neighbours reach further apart in sparse tissue than in dense
tissue — it is not the same in every cell.
Nothing here is broken. A truncated Gaussian is a perfectly respectable smoother. But if you were to write “we smoothed with a Gaussian of σ = 52 µm” in a methods section, you would be reporting a number the code never applied. That is what the warning is for, and it tells you exactly which number to quote instead:
res = ss.provenance(adata, "custom")["steps"][1]["resolved"]
res["sigma_used"] # 52.1 <- nominal; do NOT quote this
res["sigma_effective"] # 35.7 <- what the kernel behaves like; quote this
res["kernel_mass_retained"] # 0.69 <- how much of the Gaussian survived
Raise k (the default, 400, keeps ~96% of the mass) and the warning goes away, sigma_effective
converges on sigma_used, and the smoother becomes effectively fixed-bandwidth.
The persistence contract¶
Everything is in the AnnData:
key |
contents |
|---|---|
|
smoothed score |
|
unsmoothed score, same genes and combiner |
|
|
|
provenance: genes, pipeline, resolved bandwidths, version |
Write it out, and a later plotting call reads those keys. Nothing is recomputed — no kompot,
no palantir, no GP solve. That is what makes an expensive smoothing worth doing once.
import anndata as ad
adata.write_h5ad("smoothed.h5ad")
reloaded = ad.read_h5ad("smoothed.h5ad")
print("stored results:", ss.list_results(reloaded))
prov = ss.provenance(reloaded, "custom")
for step in prov["steps"]:
print(f" {step['kind']:<14} basis={step['basis']:<18} resolved={step['resolved']}")
stored results: ['composed', 'custom', 'dm_only', 'hippocampus', 'spatial_only']
kompot_gp basis=DM_EigenVectors resolved={'kompot_version': '0.8.0', 'n_landmarks_used': 5000}
knn_gaussian basis=spatial resolved={'k_used': 64, 'kernel_mass_retained': 0.6908741822395602, 'sigma_effective': 35.73044057216392, 'sigma_effective_p1': 18.98558101291319, 'sigma_effective_p99': 49.16132366135855, 'sigma_nominal': 52.10185410237582, 'sigma_used': 52.10185410237582}
# Nothing is recomputed here: the smoothed values are read straight from the file.
import time
start = time.time()
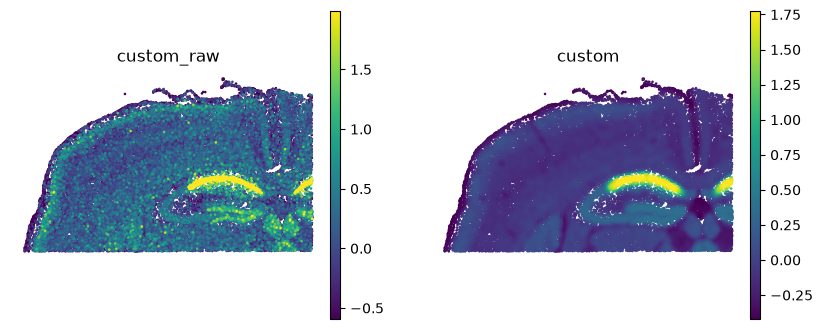
ss.pl.signature(reloaded, "custom", backend="scanpy", frameon=False)
print(f"drawing the saved result took {time.time() - start:.2f} seconds")
drawing the saved result took 0.56 seconds
Smoothing this signature took minutes. Drawing it back from the saved file took a fraction of a
second, because the field was never recomputed — spatial_smooth.plot reads obs and uns and
hands them to scanpy. That is the whole point of saving: do the expensive step once.
5. Odds and ends¶
Restrict to a subset of cells¶
Cells filtered out neither train the smoother nor receive the field. The call returns a new,
smaller AnnData — use the return value.
# A coarse annotation to filter on (this public subset ships none).
adata.obs["half"] = np.where(
adata.obsm["spatial"][:, 0] < np.median(adata.obsm["spatial"][:, 0]), "left", "right"
)
left = ss.smooth(adata, HIPPOCAMPUS, "hippocampus", subset_key="half", include=["left"])
print(f"{adata.n_obs:,} cells -> {left.n_obs:,} after the filter")
print("provenance n_obs:", ss.provenance(left, "hippocampus")["n_obs"])
36,419 cells -> 18,209 after the filter
provenance n_obs: 18209
Fit the GP on one condition, evaluate everywhere¶
KompotGP(groupby=..., condition=...) trains on one group and imputes the field for all cells —
useful when one arm of an experiment is the reference.
Other engines¶
Kde (a fine-grid FFT Nadaraya-Watson estimator, via KDEpy) renders a field rather than a
neighbour average; "spatial-gp" puts the Gaussian process on tissue coordinates with a sensible
ls_factor. Both are one steps argument away.
ss.smooth(adata, HIPPOCAMPUS, "kde", steps="spatial-kde")
ss.smooth(adata, HIPPOCAMPUS, "spatial_gp", steps="spatial-gp")
Where to go next¶
ss.provenance(adata, name)— exactly what was run, with the bandwidths it resolved.The Concepts page — composition semantics, the scoring contract, and why gene-level smoothing costs nothing in correctness.
And once more, because it is the thing that matters: these smoothed values are for looking at.
Do your statistics on adata.obs["hippocampus_raw"].